Computational Screening of Bioactive Compounds Targeting Dihydrofolate Reductase in Trypanosoma brucei for Inhibitory Activity
-
Oluokun Favour Oluwatobi
Department of Biochemistry, Faculty of Science, Lagos State University, Lagos, Nigeria
Azeez FataiDepartment of Biochemistry, Faculty of Science, Lagos State University, Lagos, Nigeria
Oladosu Micheal AbimbolaDepartment of Biochemistry, Faculty of Basic Medical Sciences, University of Lagos, Lagos, Nigeria
Moses Adondua Abah

Department of Biochemistry, Faculty of Biosciences, Federal University Wukari, Taraba, Nigeria
Igwe Ejikeme PeterDepartment of Applied Biochemistry, Faculty of Natural Sciences, Enugu State University of Science and Technology, Nigeria
Ismaila Emmanuel OluwasegunDepartment of Animal Production and Health, College of Animal Science and Livestock Production, Federal University of Agriculture Abeokuta, Ogun, Nigeria
Abiodun Wisdom OshirekuDepartment of Chemistry and Biochemistry Department, College of Physical and Mathematical Sciences, Brigham Young University, Provo, United States
Adeyeye Pius OluwaseyiDepartment of Biochemistry, Faculty of Pure and Applied Sciences, Ladoke Akintola University of Technology, Ogbomosho, Oyo, Nigeria
Akinwande Peter SaanumiDepartment of Pharmaceutical Analysis, School of Chemistry, Nottingham Trent University, United Kingdom
Ashade Noah OluwasegunResearch and Development, National Research Institute for Chemical Technology, Zaria, Nigeria
Fathia Adeola AgbolagadeDepartment of Biochemistry, Faculty of Life Sciences, Federal University of Technology Akure, Ondo, Nigeria
Nathan Rimamsanati YohannaDepartment of Biochemistry, Faculty of Biosciences, Federal University Wukari, Taraba, Nigeria
| Received 26 Mar, 2025 |
Accepted 02 Mar, 2026 |
Published 05 Mar, 2026 |
Background and Objective: Human African Trypanosomiasis is a vector-borne parasitic disease caused by Trypanosoma brucei that threatens the lives of about 60 million people globally. This condition has spread through 36 countries in Sub-Saharan Africa. The objective of this study was to assess the inhibitory potential of the bioactive compounds from plants reported for the treatment of trypanosomiasis against dihydrofolate reductase. Materials and Methods: The crystal structure of the protein was retrieved from the protein database (PDB). The structures of the phytoconstituents of the plants under study were retrieved from the PubChem database. Docking of the drug target and the phytoconstituents was carried out using PyRx software. The binding affinity energies of the compounds were assessed from docking, and their predicted pharmacokinetic and pharmacodynamic properties were assessed using DruLito and ADMETsar 2.0. Results: Some of the bioactive compounds present in Tridax procumbens, and Annona senegalensis were able to interact with the drug target with sufficient binding affinity energies (>7.4 kcal/mol), while those of Allium sativum did not bind with sufficient energy (<7.5 kcal.mol) to the drug target, and therefore did not show promise as a potential inhibitor of the drug target. Conclusion: The selected bioactive compounds present in Tridax procumbens, and Annona senegalensis have the potential to inhibit the drug target in the parasite Trypanosoma brucei and should be investigated further.
| Copyright © 2026 Oluwatobi et al. This is an open-access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |
INTRODUCTION
Dihydrofolate reductase (DHFR) is a prevalent enzyme that has wide-scale application as a drug target. A crucial function of dihydrofolate reductase (DHFR) is to catalyze the NADPH-dependent reduction of dihydrofolate to give tetrahydrofolate, a central component in the single-carbon metabolic pathway. The tetrahydrofolate is methylated to methylene tetrahydrofolate, which is directly involved in thymidine synthesis (assisting the methylation of deoxyuridine monophosphate to give thymidine monophosphate) and indirectly implicated in the metabolism of amino acids and purine nucleotide. Inhibition of DHFR thus prevents biosynthesis of DNA, leading to cell death1.
Human African Trypanosomiasis is a vector-borne parasitic disease, caused by Trypanosoma brucei, that threatens the lives of about 60 million people globally, this condition has spread through 36 countries in Sub-Saharan Africa. Nigerian medicinal plants are known to contain a large variety of chemical structures, and some of the plant extracts have been screened for antitrypanosomal activity, in the search for potential new drugs against the illness2. The disease is commonly found in riverine areas in rural settlements. However, the existing treatment for this disease is limited due to factors like the toxicity of the drugs, difficulty in the administration of drugs, and development of resistance by the parasite, among other various factors. Due to these reasons, the need for new, safe, effective, and cheap drugs are eminent and urgent3.
Bioactive compounds, naturally occurring molecules found in plants, animals, and microorganisms, have garnered significant attention in recent years due to their potential to modulate DHFR activity. The traditional use of medicinal plants with active biological compounds in the treatment and management of diseases in developing countries is on the increase as it has been shown to have potential inhibitory effects against DHFR. Findings in this area have provided evidence of the relationship between plants and medicine. Recent reports have confirmed the antitrypanosomal activities of some medicinal plants4. This research aimed at discovering inhibitors extracted from medicinal plants against the DHFR enzyme in Trypanosoma brucie. This study could provide knowledge in the design and synthesis of selective inhibitors for DHFR from protozoan parasites and also contribute to the importance of bioactive compounds as a valuable source of inspiration for drug discovery and development.
MATERIALS AND METHODS
Study area and duration: This research work was carried out between September 2023 to January 2024 at Biochemistry Laboratory, Lagos State University, Ojo Campus, Ojo Local Government, Lagos State, Nigeria.
Compound selection: Phytoconstituents of various plants that have their structures available on the PubChem database (https://pubchem.ncbi.nlm.nih.gov/) were retrieved for this study. The phytoconstituents were identified in three different plants that have been reportedly used in the treatment of Human African Trypanosomiasis (HAT), and they include Annona senegalensis, Allium sativum, and Tridax procumbens. The known substrate's molecular structure was also retrieved to compare the interaction between the plant ligands and the protein with that of the substrate and the protein; known inhibitors of the protein were also retrieved, and the interaction has been analysed to compare the interaction. All the files were downloaded in Simple Data Format (SDF).
SProtein target selection and preparation
DHFR (3QFX): An important protein implicated in folic acid metabolism, dihydrofolate reductase (DHFR), was identified as a drug target after an extensive literature review, and its crystallographic 3D structure was obtained from the protein database in CIF format. The protein was further prepared by removing heteroatoms and water molecules from the obtained molecular structure. This was carried out using a text editor (Notepad version 1.0) for removing heteroatoms to produce a nascent receptor for the ligands.
Sequence alignment: The protein sequence was run through BLAST to determine its similarity to sequences of human proteins present. The database where this operation was carried out is UNIPROT5.
Molecular docking: Before the molecular interactions between the protein and the ligand could be analyzed in silico, the files were converted into PDBQT format, which is the format recognized by the PyRx software that is used for docking. This was carried out using Open Babel version.
Molecular docking was performed using PyRx software (version 0.8.0.0), which implements AutoDock Vina as the scoring function. The grid box was centered on the active site of DHFR (PDB ID: 3QFX) with the following parameters: Center coordinates (x = 23.45, y = 15.32, z = 10.87), and grid box dimensions of 25 Å×25 Å×25 Å along the x, y, and z axes, respectively. These dimensions were selected to encompass the entire binding pocket and allow adequate conformational sampling of the ligands.
The exhaustiveness parameter was set to 8, which determines the thoroughness of the global search and represents a balance between computational time and search accuracy. AutoDock Vina's scoring function, which combines knowledge-based potentials and empirical scoring terms, was used to evaluate binding affinities. The scoring function accounts for hydrogen bonding, hydrophobic interactions, electrostatic interactions, and desolvation effects. For each ligand, the top-ranked binding pose with the most favorable binding energy (kcal/mol) was selected for further analysis. All docking simulations were performed in triplicate to ensure consistency of results.
Drug likeness screening: To ensure the ligands conform to the properties required for drug development, the molecules were screened using druLito to determine the drug-likeness property of the ligands. The SDF files of the ligand were uploaded to druLito for analysis5, and Lipinski’s rule of 5 was applied to determine the drug-likeness property.
Visualization: The docked complexes were visualized using PyMOL© Molecular Graphics System (version 2.4.0, 2010, Schrödinger LLC). The software was used to generate 3D representations of the protein-ligand complexes, visualize binding pockets, and identify the binding sites of the ligands on the protein. All molecular structure images presented in Fig. 1 and 2 were rendered using PyMOL with ray-tracing enabled for publication-quality output. The protein-ligand interaction diagrams were subsequently analyzed and annotated using the Protein-Ligand Interaction Profiler (PLIP) web server (https://plip-tool.biotec.tu-dresden.de/plip-web/plip) to identify and categorize specific molecular interactions.
Elucidating protein interaction: The PDB structure of the interacting molecules (protein/ligand) was analyzed using the Protein-Ligand Interacting Profiler (PLIP). The result of this analysis shows the various interactions available between the interacting molecules (https://plip-tool.biotec.tu-dresden.de/plip-web/plip).
Predicting admet properties: The Ligands that passed the Drug-likeness test and have binding affinity greater than 7.5 kcal/mol were analysed for the ADMET properties. The SDF files of the ligands were uploaded to the ADMETSar webserver (http://lmmd.ecust.edu.cn/) database to generate the results for its ADMET.
RESULTS
Assessment of drug-likeness of ligands: The results presented in Table 1 provide an overview of the drug-likeness characteristics of the selected ligands from Annona senegalensis and Tridax procumbens. All compounds were assessed based on key parameters such as molecular weight, lipophilicity (LogP), the number of hydrogen bond acceptors and donors, and their compliance with Lipinski’s rule of five, which is commonly used to evaluate the suitability of compounds as potential oral drugs.
| Table 1: | Drug-likeness result of the ligands | |||
| Plant source | Ligands | Molecular weight | LogP | No. of HB acceptor | No. of HB donor | No. of violations |
| Annona senegalensis | Anonaine | 265.11 | 1.809 | 3 | 1 | 0 |
| Nornantenine | 325.13 | 1.445 | 5 | 1 | 0 | |
| Tridax procumbens | Apigenin | 270.05 | 1.138 | 5 | 3 | 0 |
| Baicalein | 270.05 | 2.629 | 5 | 3 | 0 | |
| Biochanin | 284.07 | 1.364 | 5 | 2 | 0 | |
| Butein | 272.07 | 1.806 | 5 | 4 | 0 | |
| Daidzein | 254.06 | 1.124 | 4 | 2 | 0 | |
| Genistein | 270.05 | 1.043 | 5 | 3 | 0 | |
| Kaempferol | 286.05 | 1.486 | 6 | 4 | 0 | |
| Luteolin | 286.05 | 1.486 | 6 | 4 | 0 | |
| Naringenin | 272.07 | 0.79 | 5 | 3 | 0 | |
| Silymarin | 482.12 | 0.855 | 10 | 5 | 0 |
Most of the ligands, including Anonaine, Nornantenine, Apigenin, Baicalein, Biochanin, Butein, Daidzein, Genistein, Kaempferol, Luteolin, and Naringenin, have molecular weights well below the 500 Dalton threshold, suggesting favorable absorption and distribution properties. Silymarin, although having a higher molecular weight, still remains within the acceptable limit, which supports its potential as a drug candidate (Table 1).
The LogP values for all compounds are below 5, indicating that these molecules maintain a good balance between hydrophilicity and lipophilicity. This balance is essential for adequate membrane permeability and bioavailability, further supporting their potential as orally active drugs (Table 1).
Additionally, all selected ligands meet the recommended limits for hydrogen bond acceptors and donors, which are important for molecular interactions and solubility. None of the compounds recorded any violations of Lipinski’s rule of five, underscoring their favorable drug-likeness profiles.
Taken together, these findings suggest that the bioactive compounds from Annona senegalensis and Tridax procumbens possess physicochemical properties consistent with good oral bioavailability. This makes them promising candidates for further computational screening and experimental evaluation as potential inhibitors of Dihydrofolate Reductase in Trypanosoma brucei, aligning well with the objectives of this study.
Assessment of admet properties of hit ligands: Table 2 provides a detailed assessment of the ADMET characteristics of the selected bioactive compounds, or “hit ligands,” to predict their pharmacokinetic suitability and potential safety as drug candidates. These parameters are crucial in evaluating how each compound might behave in a biological system and their likelihood of success as therapeutic agents targeting Dihydrofolate Reductase in Trypanosoma brucei.
From the Table 2, it is evident that the hit ligands exhibit diverse profiles across the ADMET spectrum. For absorption, properties like blood-brain barrier (BBB) permeability, Caco-2 cell permeability, and P-glycoprotein substrate/inhibitor status are analysed. Most of the compounds show moderate to good permeability, suggesting the potential for adequate absorption if administered orally. However, a few compounds may not easily cross the BBB, which could be either beneficial or limiting, depending on the therapeutic target.
In the distribution section, parameters like plasma protein binding (PPB) and subcellular localization are considered. The table indicates that several ligands are likely to bind to plasma proteins to varying extents, which can influence their distribution and free concentration in the bloodstream. Some compounds are predicted to localize within mitochondria, which may impact their mechanism of action or toxicity.
| Table 2: | Gross composition of solvent extracted processed soyabean based diets | |||
| Class | Properties | Silymarin | Naringenin | Luteolin | Baicalein | Apigenin | ||
| ADSRPTION | BBB | NO | NO | NO | NO | NO | ||
| CaCO2 permeability | NO | NO | NO | NO | YES | |||
| Pgp-inhibitor | YES | NO | NO | NO | NO | |||
| Pgp-substrate | NO | NO | NO | NO | NO | |||
| DISTRIBUTION | PPB 5 Sub-cellular localization |
0.887758672 MITOCHONDRIA |
0.936183274 MITOCHONDRIA |
1.065997005 MITOCHONDRIA |
1.024719119 MITOCHONDRIA |
1.082861185 MITOCHONDRIA |
||
| CYP450 1A2 inhibition | NO | YES | YES | YES | YES | |||
| CYP450 3A4 inhibition | YES | YES | YES | YES | YES | |||
| CYP450 3A4 inhibition | YES | NO | NO | NO | NO | |||
| CYP450 2C9 inhibition | YES | YES | NO | NO | YES | |||
| METABOLISM | CYP450 2C9 substrate | NO | NO | NO | NO | NO | ||
| CYP450 2C19 inhibition | NO | YES | NO | NO | YES | |||
| CYP450 2D6 inhibition | NO | NO | NO | NO | NO | |||
| CYP450 2C6 substrate | NO | NO | NO | NO | NO | |||
| UGT catalyzes | YES | NO | YES | YES | YES | |||
| TOXICITY | Acute oral toxicity | Class III | Class II | Class II | Class II | Class III | ||
| hERG inhibitor | NO | NO | NO | NO | NO | |||
| Human hepatotoxicity | YES | NO | NO | NO | YES | |||
| Ames mutagenicity | NO | NO | YES | NO | NO | |||
| Carcinogens | NO | NO | NO | NO | NO | |||
| Class | Properties | Biochanin | Genistein | Butein | Daidezin | Kaempferol | Anonaine | Nornantenine |
| ADSORPTION | BBB | NO | NO | NO | NO | NO | YES | YES |
| CaCO2 permeability | YES | YES | YES | Yes | NO | NO | NO | |
| Pgp-inhibitor | NO | NO | NO | NO | NO | NO | NO | |
| Pgp-substrate | NO | NO | NO | NO | NO | NO | NO | |
| DISTRIBUTION | PPB 5 Sub-cellular localization |
1.097903132 MITOCHONDRIA |
1.09352088 MITOCHONDRIA |
0.945790052 MITOCHONDRIA |
0.965450585 MITOCHONDRIA |
0.965450585 MITOCHONDRIA |
0.871790171 MITOCHONDRIA |
0.753691316 MITOCHONDRIA |
| METABOLISM | CYP450 1A2 inhibition | YES | YES | YES | YES | YES | YES | YES |
| CYP450 3A4 inhibition | YES | YES | YES | NO | YES | NO | YES | |
| CYP450 3A4 substrate | YES | NO | NO | NO | YES | YES | YES | |
| CYP450 2C9 inhibition | YES | YES | YES | NO | YES | NO | NO | |
| CYP450 2C9 substrate | NO | NO | NO | YES | NO | NO | NO | |
| CYP450 2C19 inhibition | YES | YES | NO | YES | YES | NO | YES | |
| CYP450 2D6 inhibition | NO | NO | NO | NO | NO | YES | YES | |
| CYP450 2D6 substrate | NO | NO | NO | NO | NO | YES | YES | |
| UGT catalyzes | YES | YES | YES | YES | YES | NO | YES | |
| TOXICITY | Acute oral toxicity | Class III | Class II | Class III | Class II | Class II | Class III | Class III |
| hERG inhibition | NO | NO | NO | NO | NO | YES | YES | |
| Human hepatotoxicity | YES | YES | NO | YES | YES | NO | NO | |
| Ames mutagenicity | NO | NO | YES | NO | YES | YES | YES | |
| Carcinogens | NO | NO | NO | NO | NO | NO | NO | |
For metabolism, the interaction of ligands with major cytochrome P450 enzymes (such as CYP3A4, CYP2C9, and CYP2D6) is documented. The compounds show a range of inhibitory effects against these enzymes, which is important because strong inhibition can lead to drug-drug interactions or altered metabolism of other medications.
Excretion properties, such as renal organic cation transporter (ROCT) inhibition, are also reported for several compounds. The presence or absence of such inhibition can influence how rapidly a compound is cleared from the body.
Finally, toxicity predictions are included, with assessments such as hepatotoxicity, Ames mutagenicity, and inhibition of the hERG channel (associated with cardiac toxicity). Most of the hit ligands demonstrate low predicted toxicity, as they do not show significant hepatotoxicity or mutagenicity risks, nor do they inhibit the hERG channel appreciably.
In summary, the ADMET analysis in Table 2 suggests that while most of the selected ligands possess favorable pharmacokinetic and safety profiles, a few may have limitations that warrant further investigation. Overall, the results support the further development of these compounds as potential leads for antitrypanosomal drug discovery, with particular attention to those that combine good absorption and distribution characteristics with a low risk of toxicity or metabolic complications.
| Table 3: | Molecular interaction analysis result between the drug target and the ligands/ bioactive compounds | |||
| Plant species | Ligands | Residues involved in hydrogen bond formation and (Residues involved in π-stacking) |
Residues involved in hydrophobic interaction and (salt bridges) |
Residues involved in water bridge |
Binding affinity energy (kcal/mol) |
| Known inhibitor |
ILE(47A), ASP(88A), ARG(95B). |
-8.5 | |||
| Natural Substrate |
ILE(47A), ASP(88A), ARG(95B). |
GLY(45A), ASP(88A/88B), ARG(95B) |
-7.9 | ||
| Annona senegalensis |
Anonaine | ARG(84B), LYS(85B), ARG(107B). |
ARG(84B), LYS(85B), SER(89B, 106B) |
-8.3 | |
| Nornantenine | ASP(120B), SER(192A), LYS(229A). |
PRO(48A), LYS(93B) |
GLU(50A), LYS(123B), ASN(191A) |
-7.7 | |
| Tridax procumbens |
Apigenin | ILE(47A), SER(89A). |
SER(89A), ARG(100A) |
-8 | |
| Baicalein | ILE(47B), SER(89B). |
SER(89B) | -8.2 | ||
| Biochanin | ILE(47B), SER(89B), ARG(100B) |
SER(89B) | -8 | ||
| Butein | TRP(49B), ARG(59B) |
LEU(90B) | SER(89B) | -7.6 | |
| Daidzein | ILE(47A). SER(89A) |
SER(89A), ARG(100A) |
-7.5 | ||
| Genistein | THR(46B), ILE(47B), SER(89B), ARG(100B) |
SER(89B) | -7.8 | ||
| Kaempferol | ILE(47B, 51B), TRP(49B), SER(89B), ARG(100B) |
SER(89B) | -7.5 | ||
| Luteolin | ILE(47B), ARG(59B), SER(89B), ARG(100B) |
SER(89B, 98B), ARG(100B). |
-8.1 | ||
| Naringenin | ILE(47B). | LEU(90B). | SER(89B), PHE(94B), ARG(100B). |
-8.2 | |
| Silymarin | ASP(88A), SER(89B), ARG(95A/95B) |
TRP(49B), ASP(54B, 88B) |
-9.1 |
Table 2 presents the predicted Absorption, Distribution, Metabolism, and Toxicity (ADMET) properties of the hit ligands identified based on their strong binding affinity to the target protein (binding energy ≤ -7.5 kcal/moL). ADMET predictions were generated using ADMETsar 2.0 web server. Absorption parameters include blood–brain barrier (BBB) permeability, which indicates the ability of a ligand to cross the central nervous system barrier, Caco-2 permeability as a predictor of intestinal absorption, and P-glycoprotein (P-gp) interactions where P-gp inhibitors may enhance intracellular drug accumulation while P-gp substrates may be actively effluxed, potentially reducing bioavailability. Distribution properties include plasma protein binding (PPB), which reflects the extent of ligand binding to plasma proteins and influences the free pharmacologically active fraction, as well as predicted subcellular localization. Metabolic parameters assess potential interactions with major drug-metabolizing enzymes, including cytochrome P450 (CYP450) isoforms 1A2, 3A4, 2C9, 2C19, and 2D6, reported as either inhibitors or substrates, which are critical for predicting metabolic stability and drug-drug interaction risks. UDP-glucuronosyltransferase (UGT) catalysis indicates phase II metabolic clearance via glucuronidation. Toxicity predictions include acute oral toxicity, human Ether-à-go-go-Related Gene (hERG) inhibition as an indicator of cardiotoxicity risk, human hepatotoxicity for potential liver injury, and genotoxicity endpoints such as Ames test results, mutagenicity, and carcinogenicity. Collectively, these parameters provide an integrated assessment of the drug-likeness, pharmacokinetic behavior, and safety profile of the screened ligands.
Metabolic parameters assess potential interactions with major drug-metabolizing enzymes, including cytochrome P450 (CYP450) isoforms 1A2, 3A4, 2C9, 2C19, and 2D6, reported as either inhibitors or substrates, which are critical for predicting metabolic stability and drug-drug interaction risks. UDP-glucuronosyltransferase (UGT) catalysis indicates phase II metabolic clearance via glucuronidation. Toxicity-related predictions include acute oral toxicity, human Ether-à-go-go-Related Gene (hERG) inhibition as an indicator of cardiotoxicity risk, human hepatotoxicity for potential liver injury, and genotoxicity endpoints such as Ames test results, mutagenicity, and carcinogenicity. Collectively, these parameters provide an integrated assessment of the drug-likeness, pharmacokinetic behaviour, and safety profile of the screened ligands.
Analysis of molecular interaction between the drug target and the ligands/bioactive compounds: The molecular interaction analysis summarized in Table 3 provides insight into how the various bioactive compounds interact with the Dihydrofolate Reductase (DHFR) target in Trypanosoma brucei. The table details the specific amino acid residues involved in hydrogen bond formation, hydrophobic interactions, and water bridges or salt bridges for each ligand, along with the calculated binding affinity in terms of binding energy (kcal/mol).
The known inhibitor and natural substrate serve as reference points. The known inhibitor exhibits a strong binding affinity of -8.5 kcal/mol, interacting primarily with ILE(47A), ASP(88A), and ARG(95B) through hydrogen bonding. The natural substrate shows slightly weaker binding at -7.9 kcal/mol, with similar residues involved in both hydrogen bonding and hydrophobic interactions.
Among the compounds from Annona senegalensis, Anonaine displays a binding affinity of -8.3 kcal/mol, closely matching that of the known inhibitor. This compound primarily interacts with ARG(84B) and LYS(85B) through both hydrogen bonding and hydrophobic contacts, with additional involvement of ARG(107B) and SER(89B, 106B) through water bridges or salt bridges. Nornantenine, also from Annona senegalensis, binds with an affinity of -7.7 kcal/mol, interacting mainly with ASP(120B), SER(192A), and LYS(229A) via hydrogen bonds, while engaging PRO(48A) and LYS(93B) in hydrophobic contacts and several other residues as water bridges.
The ligands from Tridax procumbens display a range of binding affinities, generally between -7.5 and -8.2 kcal/mol, except for Silymarin, which stands out with the strongest binding affinity at -9.1 kcal/mol. Silymarin’s strong interaction is characterized by hydrogen bonds with ASP(88A), SER(89B), and ARG(95A/95B), as well as significant hydrophobic and water bridge contacts. Other notable compounds include Baicalein and Naringenin, both with binding affinities of -8.2 kcal/mol, showing interactions mainly with ILE(47B), SER(89B), and additional hydrophobic or water bridge contacts. Luteolin also demonstrates a favorable binding energy of -8.1 kcal/mol.
Overall, the results indicate that several bioactive compounds, especially Silymarin, Anonaine, Baicalein, and Naringenin, exhibit strong binding affinities and interact with key residues within the DHFR active site, similar to the known inhibitor. This suggests that these compounds have promising potential as inhibitors of DHFR in Trypanosoma brucei, supporting their further investigation and development as antitrypanosomal agents.
|
|
| Charge center aromatic ring center metal ion |
 |
| Hydrophobic interaction hydrogen bond |
|
| Water bridge π-Stacking (parallel) |
|
| π-Stacking (perpendicular) π- cation interaction halogen bond |
|
| Salt bridge Aromatic ring center |
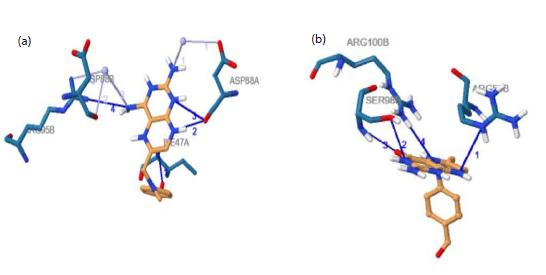
The protein-ligand interaction profiler software was used to elucidate these interactions and generate the 2D interaction diagrams. Panel A shows the binding interaction of the known inhibitor with DHFR, positioned within the enzyme’s active site and forming critical interactions with residues including ILE47A and ASP88A through hydrogen bonds and hydrophobic contacts, which stabilize the inhibitor within the binding pocket. Panel B illustrates how the natural substrate interacts with DHFR, fitting into the enzyme’s active site and forming specific interactions particularly with residues SER89A and ARG100B, which are essential for the enzyme’s normal catalytic activity. Structures were visualized and rendered using PyMOL version 2.4.0 shown in Fig 1a and b.
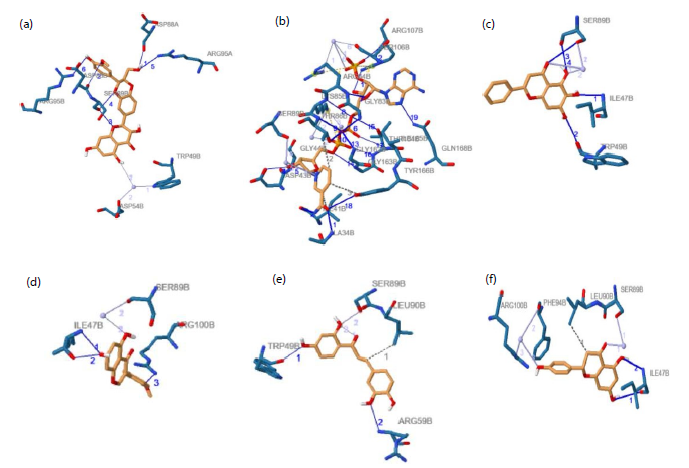
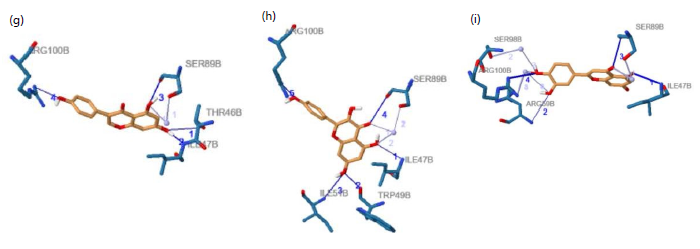
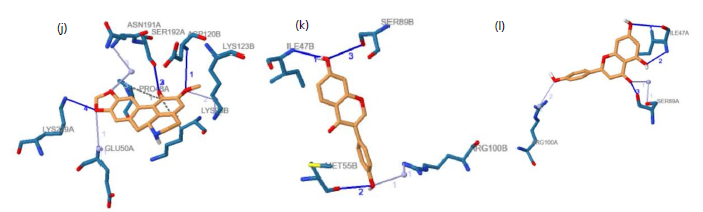
Figure 2a shows Silymarin occupying the DHFR active pocket and forming multiple stabilizing interactions with key amino acid residues, indicating strong binding stability. Figure 2b shows Anonaine interacting with active site residues through hydrogen bonding and hydrophobic contacts, supporting its favorable binding orientation. Figure 2c shows Baicalein forming specific hydrogen bonds within the catalytic region, reflecting effective positioning inside the pocket. Figure 2d shows Biochanin establishing several contacts that suggest a stable binding configuration. Figure 2e shows Butein positioned deeply within the binding site, interacting primarily through hydrogen bonds and hydrophobic effects. Figure 2f shows Naringenin adopting a compact conformation within the active cavity, engaging key residues that enhance docking stability. Figure 2g shows Genistein forming important hydrogen bonds and additional stabilizing interactions with DHFR. Figure 2h shows Kaempferol interacting at multiple binding points within the active site, indicating a strong binding pose. Figure 2i shows Luteolin establishing significant interactions with essential residues, highlighting its inhibitory potential. Figure 2j shows Nornantenine forming both hydrogen bonds and hydrophobic contacts within the pocket. Figure 2k shows Daidzein fitting appropriately into the binding cavity and forming stabilizing interactions. Figure 2l shows Apigenin forming targeted hydrogen bonds within the active site, suggesting structural compatibility and potential inhibitory activity.
DISCUSSION
The HAT is a disease caused by the protozoan parasite Trypanosoma brucei. The existing therapies for this otherwise fatal condition are limited due to its toxicity, emerging resistance, difficulty in administration, and the cost of treatment6. This study investigate the potential of phytoconstituents from herbs used traditionally to treat this condition to inhibit a drug target in the parasite, thereby becoming toxic and eliminating the parasite.
A very important protein found in the parasite that has been proven to be a good drug target is DHFR. This protein is responsible for folate metabolism in the parasite7. The affinity of sthe natural substrate and the known inhibitor to the drug target was analyzed and used to set a baseline affinity value of -7.5 kcal/mol for the ligands analyzed in this study. The ligands that passed the first stage of screening were then tested for drug-likeness properties using druLito software8. Afterwards, the ligands that passed the drug-likeness screening were then uploaded to a profiler database, where the interaction between the ligand and the drug target was analyzed by uploading the PDB file of the interacting molecules (generated using PyMol software) and the result showed the chain(s) and amino acid residues involved in the interaction9,10.
The ligands, Anonaine and Nornantenine, of the four ligands under observation from Anona senegalensis passed the screening stages stated above and also bind to the drug target around the binding site of the natural substrate and the known inhibitor, indicating they have the potential of inhibiting the protein by altering the conformation of the active site allosterically11-13. While the ligands: Apigenin, Baiclein, Biochanin, Butein, Geinstein, Kaempferol, Luteolin, Naringenin, and Sylimarin from the plant Tridax procumbens have the potential to act as effective inhibitors for the drug target, due to their high binding affinity for the ligand and bind to residues at sites similar to that of the natural substrate and the known inhibitor (as observed in silymarin) or close to the residues as observed in the other Protein-Ligand interactions14,15. Of all the plants used in the course of this study, Allium sativum ligands exhibited a very low binding affinity for the drug target and therefore did not show any promise in inhibiting the drug target, and of all the ligands used in this study, silymarin showed the greatest potential in inhibiting the drug target, due to its high binding affinity value of -9.1 kcal/mol, which is greater than the binding affinity value of the natural substrate and the known inhibitor. It showed greater inhibitory potential when the protein-ligand interaction was observed using protein-ligand interaction profiler (PLIP), the results showed that it shares interaction with 2 residues (88A-ASP and 95B-ARG) as the known inhibitor of the drug target, and also shares one amino acid residue (88A-ASP) with the natural substrate16,17. Silymarin is a flavoligand present in plants such as Tridax procumbens. Silymarin has been shown to possess various pharmacological properties like hepatoprotective, antioxidant, anti-inflammatory, anticancer, and cardioprotective activities18,19. Silymarin is widely used in treating hepatic dysfunction due to its pharmacological functions, but its activity against Trypanosomiasis is yet to be investigated20. This study is limited by its reliance on in silico molecular docking and ADMET predictions without experimental validation, which may not fully reflect the biological activity and pharmacodynamics of the identified compounds against Trypanosoma brucei. Future studies should include in vitro enzyme inhibition assays, cell-based antitrypanosomal evaluations, and molecular dynamics simulations to confirm binding stability and validate the therapeutic potential of the identified DHFR inhibitors.
CONCLUSION
Computational screening identified several promising inhibitors of Trypanosoma brucei dihydrofolate reductase (TbDHFR), a critical enzyme in the parasite’s folate metabolism. Selected compounds demonstrated favorable binding affinities and acceptable drug-likeness and ADMET profiles, with silymarin showing the strongest interaction. These findings highlight structurally diverse natural compounds as potential lead candidates for antitrypanosomal drug development. However, experimental validation is required to confirm their biological efficacy and safety.
SIGNIFICANCE STATEMENT
Human African Trypanosomiasis, caused by Trypanosoma brucei, remains a major neglected tropical disease with limited safe treatment options. This study identifies plant-derived compounds targeting TbDHFR as potential therapeutic leads using a cost-effective computational approach. The findings provide a scientific basis for further experimental development of novel antitrypanosomal agents.
REFERENCES
- Hoare, C.A., 1972. The Trypanosomes of Mammals: A Zoological Monograph. Blackwell Scientific Publications, Oxford, United Kingdom, ISBN: 978-0632082001, Pages: 749.
- Lipinski, C.A., 2008. Compound Properties and Drug Quality. In: The Practice of Medicinal Chemistry, Wermuth, C.G. (Ed.), Elsevier, Amsterdam, Netherlands, ISBN: 978-0-12-374194-3, pp: 481-490.
- Nok, A.J., S. Williams and P.C. Onyenekwe, 1996. Allium sativum-induced death of African trypanosomes. Parasitol. Res., 82: 634-637.
- Nwodo, N.J., A. Ibezim, F. Ntie-Kang, M.U. Adikwu and C.J. Mbah, 2015. Anti-trypanosomal activity of Nigerian plants and their constituents. Molecules, 20: 7750-7771.
- Oscherwitz, S.L., 2003. East African trypanosomiasis. J. Travel Med., 10: 141-143.
- Ponnusamy, S., R. Ravindran, S. Zinjarde, S. Bhargava and A.R. Kumar, 2011. Evaluation of traditional Indian antidiabetic medicinal plants for human pancreatic amylase inhibitory effect in vitro. Evidence-Based Complementary Altern. Med., 2011.
- Priya, L.B., R. Baskaran and V.V. Padma, 2017. Phytonanoconjugates in Oral Medicine. In: Nanostructures for Oral Medicine, Andronescu, E. and A.M. Grumezescu (Eds.), Elsevier, Amsterdam, Netherlands, ISBN: 978-0-323-47720-8, pp: 639-668.
- Kennedy, P.G.E., 2004. Human African trypanosomiasis of the CNS: Current issues and challenges. J. Clin. Invest., 113: 496-504.
- Robays, J., A.E. Kadima, P. Lutumba, C.M. mia Bilenge and V.K.B.K. Mesu et al., 2004. Human African trypanosomiasis amongst urban residents in Kinshasa: A case-control study. Trop. Med. Int. Health, 9: 869-875.
- Priotto, G., S. Kasparian, W. Mutombo, D. Ngouama and S. Ghorashian et al., 2009. Nifurtimox-eflornithine combination therapy for second-stage African Trypanosoma brucei gambiense trypanosomiasis: A multicentre, randomised, phase III, non-inferiority trial. Lancet, 374: 56-64.
- Seixas, J.B.A., 2004. Investigations on the Encephalopathic Syndrome During Melarsoprol Treatment of Human African Trypanosomiasis. Verlag Nicht Ermittelbar, Universidade NOVA de Lisboa, Portugal, Pages: 249.
- Kennedy, P.G.E., J. Rodgers, B. Bradley, S.P. Hunt and G. Gettinby et al., 2003. Clinical and neuroinflammatory responses to meningoencephalitis in substance P receptor knockout mice. Brain, 126: 1683-1690.
- Lejon, V., J. Lardon, G. Kenis, L. Pinoges and D. Legros et al., 2002. Interleukin (IL)-6, IL-8 and IL-10 in serum and CSF of Trypanosoma brucei gambiense sleeping sickness patients before and after treatment. Trans. R. Soc. Trop. Med. Hyg., 96: 329-333.
- Mesu, V.K.B.K., W.M. Kalonji, C. Bardonneau, O.V. Mordt and S. Blesson et al., 2018. Oral fexinidazole for late-stage African Trypanosoma brucei gambiense trypanosomiasis: A pivotal multicentre, randomised, non-inferiority trial. Lancet, 391: 144-154.
- Solano, P., V. Jamonneau, P. N'Guessan, L. N'Dri and N.N. Dje et al., 2002. Comparison of different DNA preparation protocols for PCR diagnosis of human African trypanosomosis in Côte d'Ivoire. Acta Trop., 82: 349-356.
- Chappuis, F., L. Loutan, P. Simarro, V. Lejon and P. Büscher, 2005. Options for field diagnosis of human African trypanosomiasis. Clin. Microbiol. Rev., 18: 133-146.
- Donelson, J.E., 2003. Antigenic variation and the African trypanosome genome. Acta Trop., 85: 391-404.
- Kennedy, P.G.E., 2006. Diagnostic and neuropathogenesis issues in human African trypanosomiasis. Int. J. Parasitol., 36: 505-512.
- Kennedy, P.G.E., 2013. Clinical features, diagnosis, and treatment of human African trypanosomiasis (sleeping sickness). Lancet Neurol., 12: 186-194.
- Franco, J.R., G. Cecchi, G. Priotto, M. Paone and A. Diarra et al., 2017. Monitoring the elimination of human African trypanosomiasis: Update to 2014. PLoS Negl. Trop. Dis., 11.
How to Cite this paper?
APA-7 Style
Oluwatobi,
O.F., Fatai,
A., Abimbola,
O.M., Abah,
M.A., Peter,
I.E., Oluwasegun,
I.E., Oshireku,
A.W., Oluwaseyi,
A.P., Saanumi,
A.P., Oluwasegun,
A.N., Agbolagade,
F.A., Yohanna,
N.R. (2026). Computational Screening of Bioactive Compounds Targeting Dihydrofolate Reductase in Trypanosoma brucei for Inhibitory Activity. Research Journal of Parasitology, 21(1), 9-12. https://doi.org/10.3923/parasite.2026.09.12
ACS Style
Oluwatobi,
O.F.; Fatai,
A.; Abimbola,
O.M.; Abah,
M.A.; Peter,
I.E.; Oluwasegun,
I.E.; Oshireku,
A.W.; Oluwaseyi,
A.P.; Saanumi,
A.P.; Oluwasegun,
A.N.; Agbolagade,
F.A.; Yohanna,
N.R. Computational Screening of Bioactive Compounds Targeting Dihydrofolate Reductase in Trypanosoma brucei for Inhibitory Activity. Res. J. Parasitol 2026, 21, 9-12. https://doi.org/10.3923/parasite.2026.09.12
AMA Style
Oluwatobi
OF, Fatai
A, Abimbola
OM, Abah
MA, Peter
IE, Oluwasegun
IE, Oshireku
AW, Oluwaseyi
AP, Saanumi
AP, Oluwasegun
AN, Agbolagade
FA, Yohanna
NR. Computational Screening of Bioactive Compounds Targeting Dihydrofolate Reductase in Trypanosoma brucei for Inhibitory Activity. Research Journal of Parasitology. 2026; 21(1): 9-12. https://doi.org/10.3923/parasite.2026.09.12
Chicago/Turabian Style
Oluwatobi, Oluokun, Favour, Azeez Fatai, Oladosu Micheal Abimbola, Moses Adondua Abah, Igwe Ejikeme Peter, Ismaila Emmanuel Oluwasegun, Abiodun Wisdom Oshireku, Adeyeye Pius Oluwaseyi, Akinwande Peter Saanumi, Ashade Noah Oluwasegun, Fathia Adeola Agbolagade, and Nathan Rimamsanati Yohanna.
2026. "Computational Screening of Bioactive Compounds Targeting Dihydrofolate Reductase in Trypanosoma brucei for Inhibitory Activity" Research Journal of Parasitology 21, no. 1: 9-12. https://doi.org/10.3923/parasite.2026.09.12

This work is licensed under a Creative Commons Attribution 4.0 International License.